Standard protocol Toshiyuki Hatano 070525
How to construct the expression system of Green fluorescent fusion protein in E. coli ver.8
Outline
A method to fuse fluorescent protein (GFP, YFP, CFP) to N or C terminal of a protein of interest. The advantage of this method is that fusion proteins are expressed under the control of its native promoter.
Apply the homologous recombination system established by Wanner et al., 2000, PNAS, p6640 and Court et al., 2000, PNAS 97 p9597 to insert a DNA fragment encoding a fluorescent protein into target sequence.
Schematic representation of this method is shown as follows.
Figure (Hatano et al., 2007, 64, Mol. Microbiol)
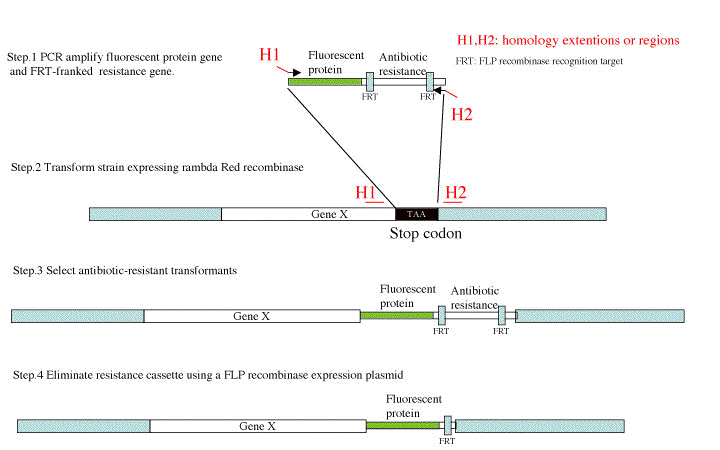
2. Preparation of a DNA fragment(For C terminal fusion, Figure Step.1)
2-2-1 Design of primers to prepare a DNA fragment
Choose 40b sequences outside of the stop codon indicated as H1, H2 in Figure Step2. H2 is a complementary sequence for PCR primer.
Add H1 to 20bp sequence from first Met codon of gfpuv4* (GFP), eyfp (YFP) and ecfp (CFP).
Add H2 to Priming site1 (P1) sequence (determined by Wanner et al.) indicated as right allow in Figure Step1.
(1) Direct fusion to C-terminal (no linker)
EG1: Gene X-GFPuve4 fusion
GeneX-gfp-U
H1+ atgagtaaaggagaagaact(20bp from first Met codon of gfpuv4)
GeneX-D
H2+ gtgtaggctggagctgcttc(priming site 1)
EG2: GeneX-EYFP fusion. The same primers are applicable to ECFP, Venus
GeneX-yfp-U
H1+ gtgagcaagggcgaggagct(20bp next to first Met codon of EYFP)
GeneX-D
H2+ gtgtaggctggagctgcttc(priming site 1)
Illustrative case: SopA-GFPuv4 fusion (Hatano et al., 2007 Mol. Microbiol, 64, 1198-1213)
(SD sequence is added to after H2 sequence down stream primer)
 SSopAdelstop(C)GFP SSopAdelstop(C)GFP
aattttcgatcgtctgattaaaccacgctgggagattaga atgagtaaaggagaagaact
upstream of stop codon of SopA GFPuv4*
  ASSopAdelstopSD(C)Kmp1 ASSopAdelstopSD(C)Kmp1
tattgagcgtatgttttggaataacaggcgcacgcttcat tttattatctccc gtgtaggctggagctgcttc
downstream of stop codon of SopA SD priming site (p1)
(2)Fusion with a linker
Addition of 4xGlycine linker between GeneX and GFP
a) Amplify a DNA fragment by PCR with primers as follows using a template described bellow (Template Preparation)
4xgly-GFP
ggaggaggaggaagtaaaggagaagaact (firstMetから20bp)
P1
gtgtaggctggagctgcttc
b) Amplify a DNA fragment with with primers as follows using the PCR product obtained in a) as a template.
GeneX-U
H1+GGAGGAGGAGGAAGTAAAGG(+20bp)
GeneX-D
H2+ gtgtaggctggagctgcttc(priming site 1)
2-2-2 Preparation of Screening Prinmers
Prepare a set of primers 60bp upstream sequense or complimentary seaquence of 60bp downstream of the X-gene to test whether the DNA fragment is inserted to the target sequence.
EG:
geneX-60-u
sequence of 20-30bp upstream of geneX
geneX-60-d
complementary sequence of 20-30bp down stream of geneX
2-2-3 Preparation of Template
Linearize the following plasmid
GFP; pTH5 (pGFPuv4*kan5) Strain:TH5 (DH10B/pTH5)、
YFP; pTH64 (pEYFP-FRT-kan-FRT) Strain:TH64 (DH10B/pTH64)、
CFP; pTH59 (pECFP-FRT-kan-FRT) Strain:,TH59 (DH10B/pTH59)、
Venus; pTH1017(pCS2/Venus-FRT-kan-FRT) Strain:TH1017 (DY330/pTH1017:Ts)、
Procedure;
Digest pTH5, pTH1017, pTH64, pTH59 with HindIII, extract and column purify single band of DNA in agarose electrophoresis gel. Dilute DNA with 100 μl of 1/10TE.
2-3 PCR Reaction
Amplify 2.3kb DNA fragment using 1/1, 1/50, 1/100 diluted solution obtained in 2-2 in a PCRcondition as follows.
The template should be diluted as much as possible to avoid appearance of Kanamycin resistant transformant caused by undigested plasmids.
PCR condition
(Pyrobest, TAKARA)
Pyrobest buffer II 20μl
2.5mM dNTP mixture 16μl
template 2μl
primer 10μM 4μl
10μM 4μl
ΔmilliQ 152μl
Pyrobest polymerase 1μl
Divide reaction solution into 25μl solution in a tube (x8) to reduce mutation frequency.
30 x (98°C: 10sec
57°C: 30sec
68°C: 2min)
72°C 7min
Purify PCR product with Concert Rapid PCR purification kit
and dilute with 50μl of 1/10TE
Subject 2μl of the PCR product in agarose gel electrophoresis
Single 2.2kb band should be detected under EtBr staining.
3 Integration of DNA fragment(Figure Step. 2, 3, 4)
3-1 Electroporation of the DNA fragment into the strains for homologous recombination(Figure Step. 2, 3)
If the target gene is on chromosome, transform a strain by pKD46 (Wanner et al.) and induce Red recombination system with arabinose, then prepare as competent cell for electroporation.
If the target gene is on plasmid, transform DY330 (Court et al.), or DY331 (pBR origin plasmid) by the plasmid and induce Red recombination system with 42C° heat shock , then prepare as competent cell for electroporation.
In each case, electroporate 1-2μl of the PCR product prepared in 2-3 into 20μl of the competent cell with 2.44kV (or above) pulse.
↓
Strain with pKD46 should be incubated in SOB 1ml at 25°C over night.
DY330/plasmid should be grown in L medium with selection marker for the plasmid.
↓
Selection of antibiotic resistant transformants and PCRscreening (Figure Step.3)
(Selection with antibiotics)
Spread concentrated cells on 15-20μg/ml kanamycin L plate and incubate at 37 °C for strain/pKD46 or at 30°C DY330over night.
↓
Single colonization
Separate single cell by streaking at least 8 independent colonies thinly using 3 toothpicks on 15-20μg/ml kanamycin L plate and incubate them over night.
This procedure should never be omitted to avoid getting false positive candidate.
Otherwise screening efficiency will be extremely low.
3-2 Screening of recombinant (Figure Step. 3)
Check insertion of the DNA fragment by colony PCR using the Screening Prinmers 2-2-2.
(criteria)
Length of Xgene: xkb,
Length of Xgene with inserted fragment : x+2.3kb
PCR conditon (LA Taq)
LA Taq 10xbuffer 10μl
2.5mM dNTP Mix 10μl
25mM MgCl2 10μl
primer 10μM geneX-60-u 2μl
geneX-60-d 2μl
ΔmilliQ 66μl
LA Taq 0.5μl
Gently mix the reaction mixture and divide 10μl in reaction tube.
Pick up single colony by toothpick and streak cells on Km Lplate and then dip in the reaction solution.
Use parent strain or strain with parent plasmid as negative controle
94°C 2min.
30x (98°C : 10sec.,
57°C : 10sec.,
68°C : 2-4min)
72°C : 7min
4°C
Subject 5μl of PCR product in agarose gel electropholesis and check whether the size of the band mach the criteria.
3-3 Elimination of FRT kanamycin resistant cassette by FLP (Figure Step. 4)
(Introduction of pCP20)
For chromosomal integration, Electroporate pCP20 into the positive clone in 3-2.
For plasmid, extract the plasmids from positive clone n 3-2 and then electroporate the obtained plasmid in to the strain harboring pCP20 (EG. AB1157/pCP20).
↓
SOB1ml (Cm15μg/mlL for plasmid)
30°C, 1.5hr incubate
↓
spread on Cm15μg/ml+Km15μg/ml (add more antibiotics for plasmids) L plate, incubate at 30°C over night.
↓
Separate single cell by streaking at least 8 independent colonies thinly using 3 toothpicks on Cm15μg/ml+Km15μg/ml (add more antibiotics for plasmids) L plate and incubate them at 42°C over night.
3-3-1 Selection of clones in which the FRT cassette iseliminated
Replicate independent colonies on each L (with antibiotics for plasmid), Km15μg/ml, Cm15μg/ml and incubate at 37°C over night.
Select strain only grown on L (with antibiotics for plasmid), but neghther on Km15μg/ml L nor Cm15μg/ml L plate.
3-3-2 Confirmation of elimination of FRT kanamycin resistant cassette by PCR.
Check whether the FRT cassette is eliminated by colony PCR of the Km and Cm sensitive candidate obtained in 3-3-1 using the Screening Prinmers.
(criteria)
GeneX: xkb
GeneX+fluorescent protein gene : x+0.7kb
PCRcondition (LATaq)
LA Taq 10xbuffer 10μl
2.5mM dNTP Mix 10μl
25mM MgCl2 10μl
primer 10μM geneX-60-u 2μl
geneX-60-d 2μl
ΔmilliQ 66μl
LA Taq 0.5μl
Gently mix the reaction mixture and divide 10μl in reaction tube.
Pick up single colony by toothpick and streak cells on Km Lplate and then dip in the reaction solution.
Use parent strain or strain with parent plasmid as negative controle
94°C 2min.
30x (98°C 10sec.
57°C 10sec.
68°C 2-4min)
72°C 7min
4°C
Subject 5μl of PCR product in agarose gel electropholesis and check whether the size of the band mach the criteria.
(Check fluorescence)
Observe fluorescent signal under the conventional fluorescent microscopy)
(Expression of the protein)
Check the size of fusion protein (Molecular Weight of the target protein +27kDa) by immunoblot analysis using anti GFP antibody (A.v. peptide antibody, Clontech)
Standard procedure is the same as described above for C terminal fusion.
The different point in Template and Primers are described below.
pTH36 template plasmid for N-terminal fusion of GFPuv4 gene.
To generate “gggaagttcctattctctagaaagtataggaacttc” encoding a linker “GKFLFSRKYRNF” amino acid sequence by eliminating the FRT kan cassette, “gg” was added to the sequence “gaagttcctattctctagaaagtataggaacttc” in FRT sequence in Wanner et al.
I.O.W.,
GFP (Δstop codon)-(the modified FRT (compliment)-GeneX
.....gcatggatgagctctacaaa-gggaagttcctattctctagaaagtataggaacttc-sequence of geneX
EG: Gfpuv4-GeneX fusion protein
GeneX-gfp-U
H1 (upstream of firstMet of GeneX)+ atgagtaaaggagaagaact (20bp of gfpuv4 from first Met codon)
GeneX-D
H2 (downstream of firstMet of GeneX)+ gaagttcctatactttc (for modified FRT sequence)
Illustrative case: GFPuv4-Glyx4-SopA
 SSopA(N)GFPuv SSopA(N)GFPuv
aataaaaaaacgcaaaggcaatgattaaaggatgttcaga atgagtaaaggagaagaact
upstream of SopA GFPuv4*
  ASd)SopA(delMet)(comp)3’ ASd)SopA(delMet)(comp)3’
gaccagcgtttatgcactggttaagtgtttccatgagttt tcctcctcctcc gaagttcctatactttc
SopAΔfirstMet Gly*4 modified FRT
Others
YFP N-terminal fusion: pTH380
CFP N-terminal fusion: pTH383 |
